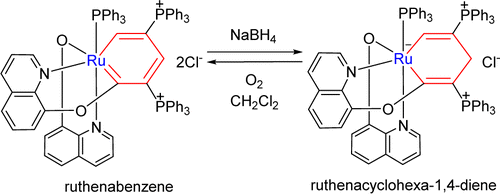
Why Does Activation of the Weaker C═S Bond in CS2 by P/N-Based Frustrated Lewis Pairs Require More Energy Than That of the C═O Bond in CO2? A DFT Study
Submitted by Jun Zhu on Tue, 12/09/2014 - 23:30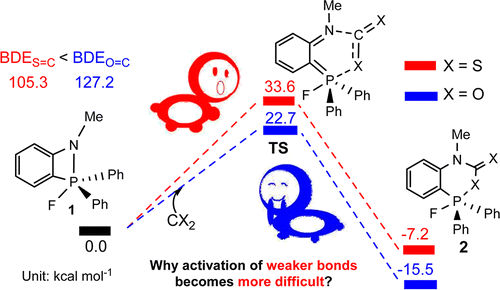
The sequestration of carbon disulfide (CS2), a common pollutant in environmental systems, is of great importance due to its physical harm to human beings. Compared with CO2 capture, that of CS2 is much less developed. The use of P/N-based frustrated Lewis pairs (FLPs) has been proven, both experimentally and theoretically, to be an alternative strategy to efficiently sequestrate CO2. Therefore, we pose the question of whether the analogue CS2 could also be sequestrated by the same FLPs, given that the C═S bond in CS2 is weaker than the C═O bond in CO2.