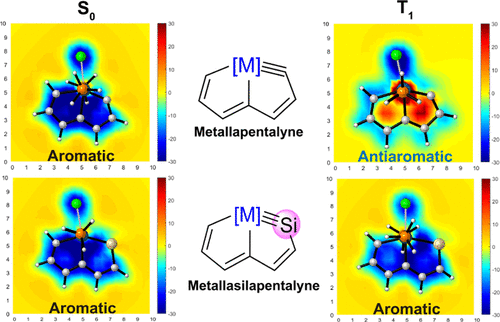
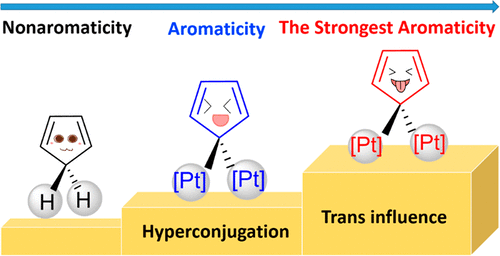
Predicting the Hyperconjugative Aromaticity in Cyclopentadiene Containing Group 8 Transition Metal Substituents
Submitted by Jun Zhu on Wed, 09/18/2024 - 08:23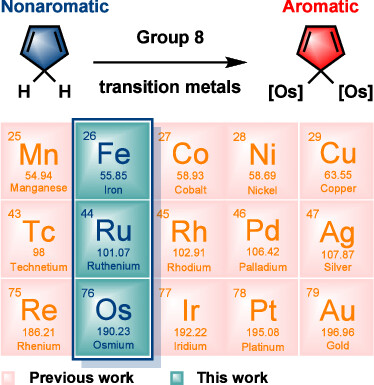
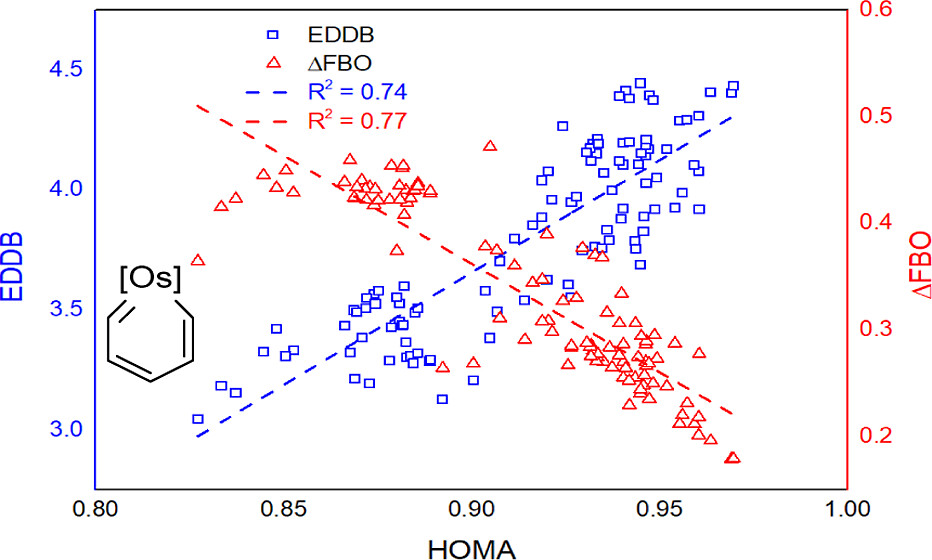
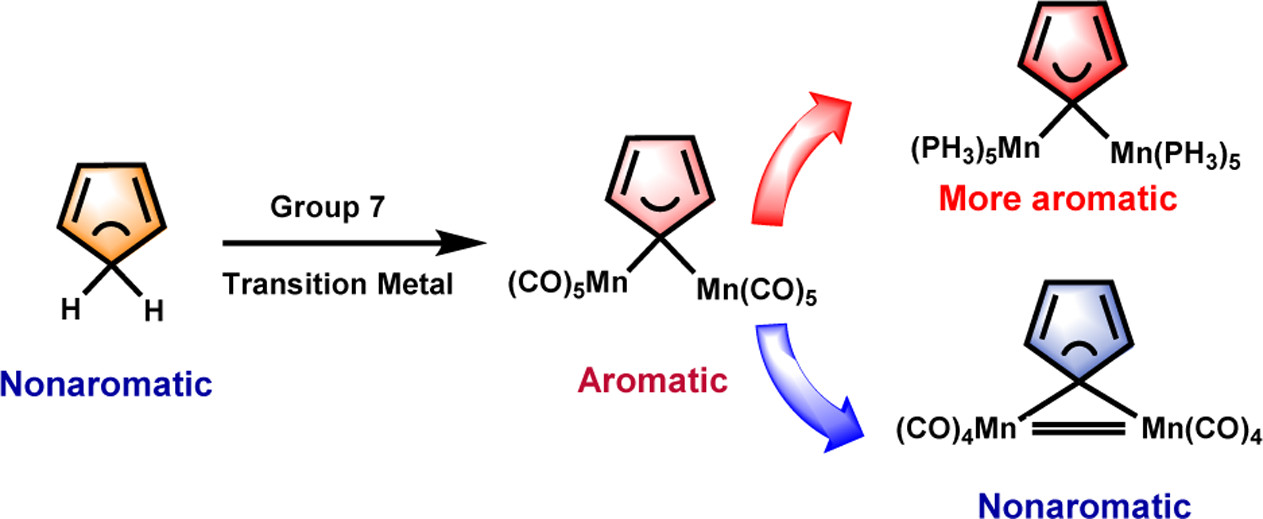
Hyperconjugative aromaticity, an integration of two chemical concepts, aromaticity and hyperconjugation, was first proposed in 1939. Recently, breaking through the main group elements, the hyperconjugative aromaticity has been successfully extended to the transition metal system, including groups 7, 9, 10, and 11 organometallic substituents. Here, we demonstrate that the missing group 8 transition metal substituents also possess a powerful ability to induce hyperconjugative aromaticity in cyclopentadiene via density functional theory calculations.