Theoretical study on the interconversion of silabenzenes and their monocyclic non-aromatic isomers via the [1,3]-substituent shift: Interplay of aromaticity and Bent's rule
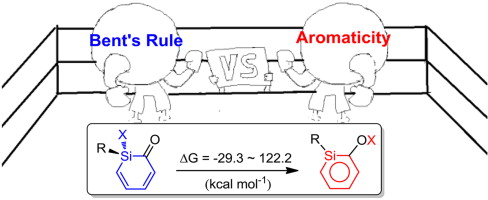
Density functional theory (DFT) calculations were performed to examine the substituent effects on the interconversion of silabenzenes and their monocyclic non-aromatic isomers. A previous study suggested that aromaticity is the driving force for this process. Interestingly, our systematic calculations reveal that the contribution from aromaticity can be evaluated quantitatively (ca. 30 kcal mol-1). Thus it is the interplay of aromaticity and Bent's rule that determine their relative stabilities. In addition, strong correlations of reaction energies as well as reaction barriers have been identified against the s character of Si in the Si-X bonds, providing an important guide to the synthesis of silabenzenes.
http://www.sciencedirect.com/science/article/pii/S0022328X14004021