Charge-driven stability and aromaticity of C₂N₂B₂H₄ isomers: Insights from a combined DFT and machine learning study
Submitted by Jun Zhu on Mon, 04/14/2025 - 08:23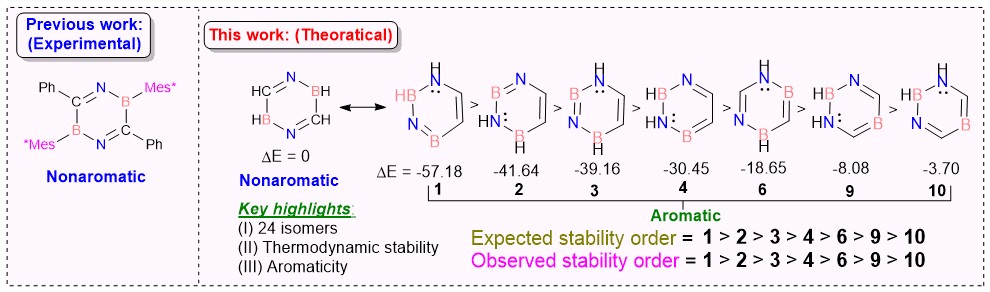
Recent research has sparked significant interest in exploring the effects of BN unit doping on the electronic structure of isoelectronic and isostructural benzene analogs, driven by their promising applications in pharmaceuticals and material sciences. In this study, we provide the first comprehensive investigation of BN/CC isosterism in 2,5-dihydro-1,4,2,5-diazadiborinine and its isomers (1−13) through density functional theory (DFT) calculations and machine learning-based analysis.