Charge-driven stability and aromaticity of C₂N₂B₂H₄ isomers: Insights from a combined DFT and machine learning study
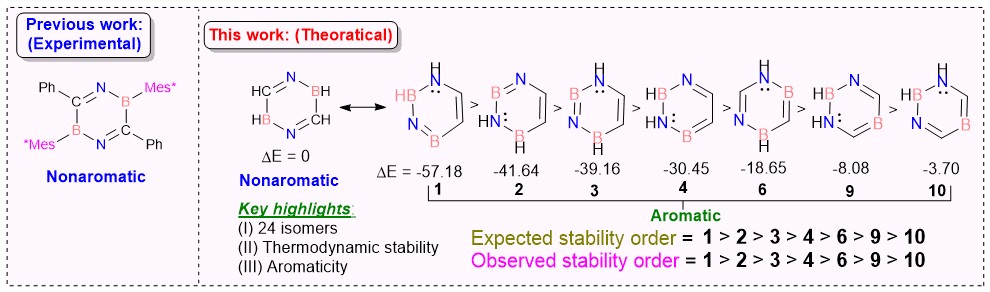
Recent research has sparked significant interest in exploring the effects of BN unit doping on the electronic structure of isoelectronic and isostructural benzene analogs, driven by their promising applications in pharmaceuticals and material sciences. In this study, we provide the first comprehensive investigation of BN/CC isosterism in 2,5-dihydro-1,4,2,5-diazadiborinine and its isomers (1−13) through density functional theory (DFT) calculations and machine learning-based analysis. Our findings reveal that isomers featuring jointed N−B−N−B linkages (1−4) exhibit the highest thermodynamic stability, attributed to alternatively distributed charge across the ring, facilitating enhanced electron delocalization. In contrast, isomers with disjointed B and N atoms, or those containing B−B and N−N bonds, exhibit greater charge separation, leading to reduced stability (5−12). Notably, 13 is the least stable due to its non-planarity and disrupted conjugation. Natural Resonance Theory (NRT) analysis confirms that the most stable isomers from three distinct categories (1, 5, and 11) form benzyne-like structures, characterized by triple bonding between N and B. Aromaticity evaluation using NICS(1)zz, HOMA, EDDBπ, and MCI indexes confirms that 1–12 exhibit moderate to high aromaticity in the singlet state (S₀), whereas 13 is nonaromatic. To evaluate the relative contributions of different aromaticity descriptors, Principal Component Analysis (PCA) was performed, revealing that NICS(1)zz is the most reliable predictor of aromaticity. Additionally, iterative linear regression modeling and PCA were used to determine the primary factors governing thermodynamic stability. Our results conclusively identify NPA charge distribution as the most significant predictor of stability, while bond order and aromaticity are less important. These insights provide a quantitative framework for understanding BN-heterocycle stability, offering valuable design strategies for tailoring the electronic properties of novel heterocyclic compounds for targeted applications in materials science and drug discovery.
https://pubs.rsc.org/en/Content/ArticleLanding/2025/OB/D5OB00131E