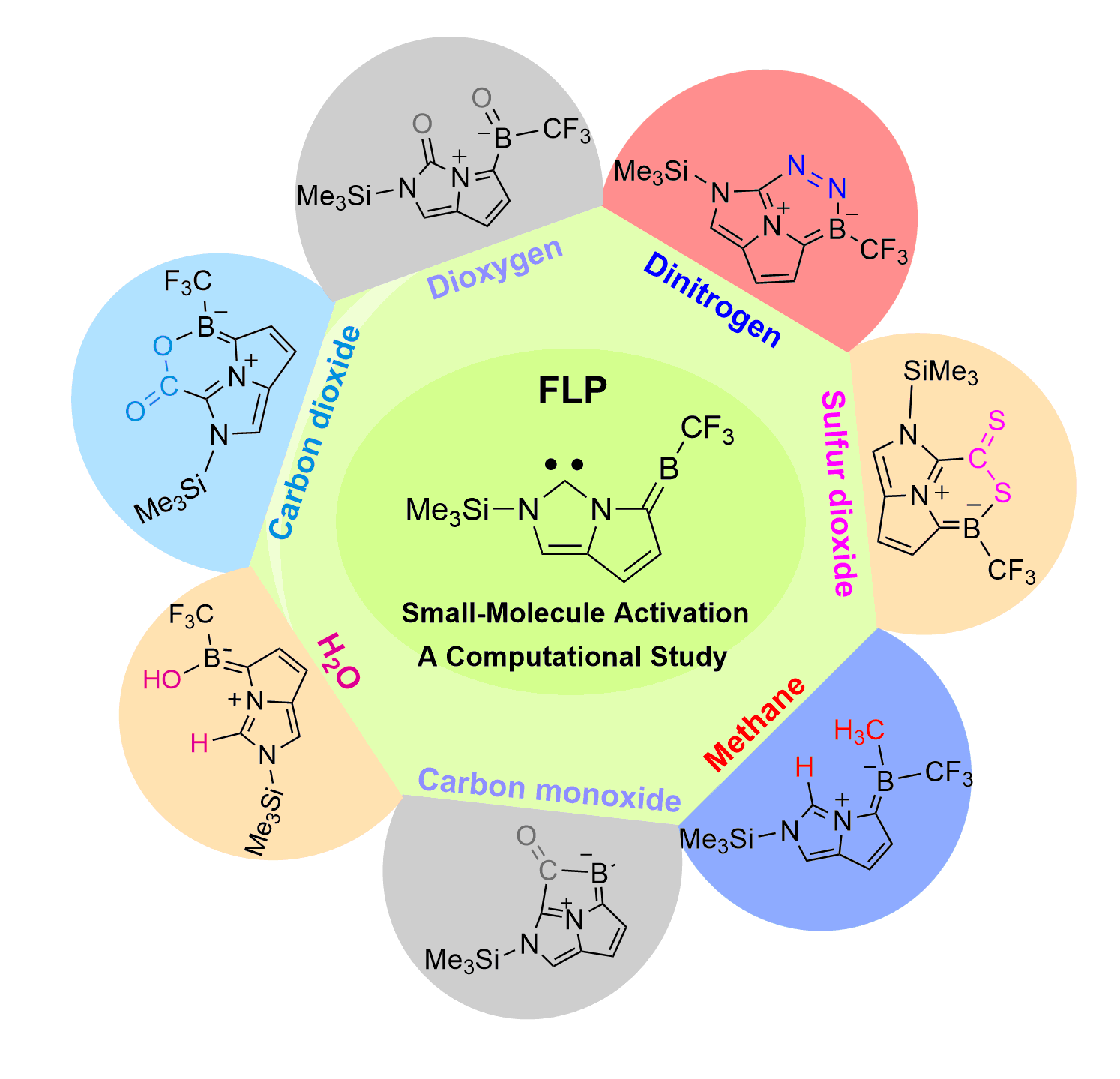
Electrochemical cascade migratory versus ortho-cyclization of 2-alkynylbenzenesulfonamides
Submitted by Jun Zhu on Wed, 01/24/2024 - 15:16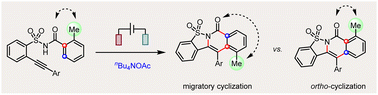
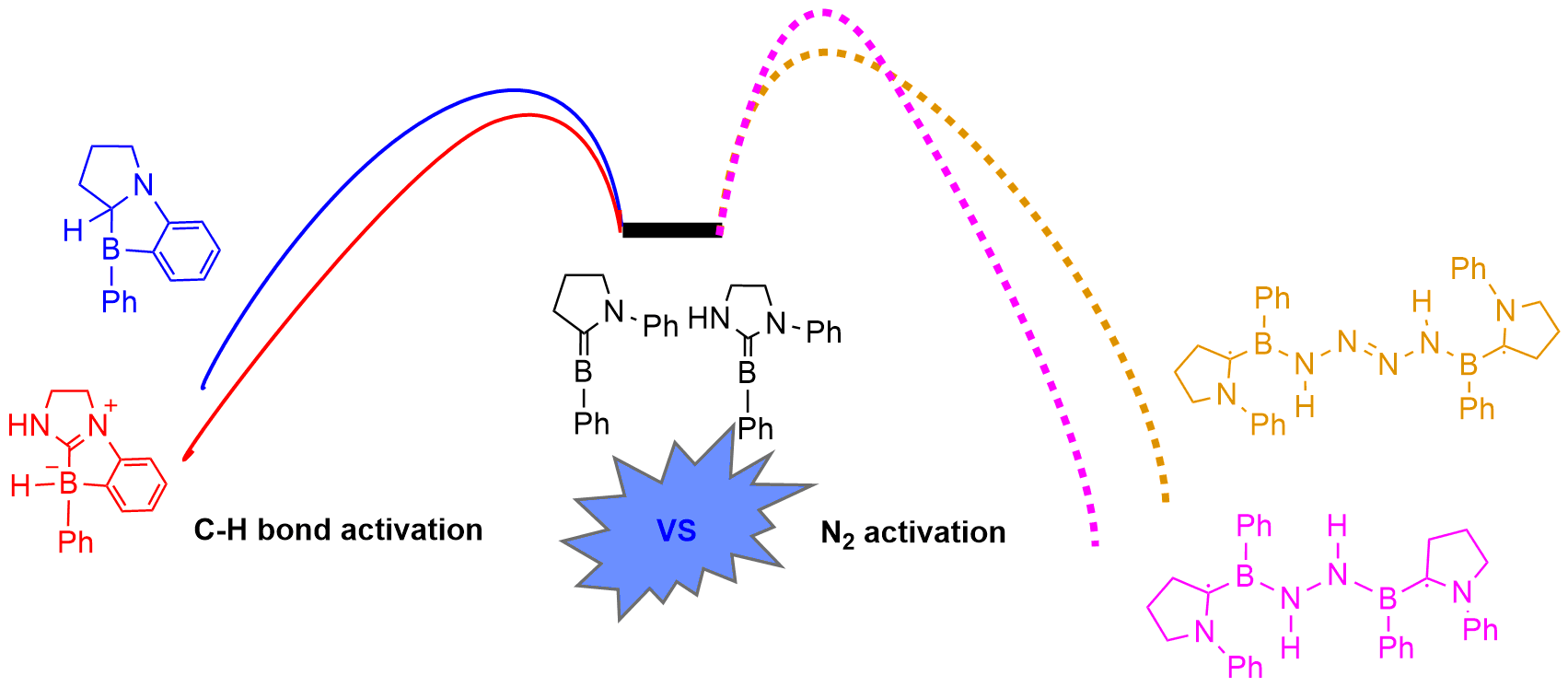
Efficient control over several possible reaction pathways of free radicals is the chemical basis of their highly selective transformations. Among various competing reaction pathways, sulfonimidyl radicals generated from the electrolysis of 2-alkynylbenzenesulfonamides undergo cascade migratory or ortho-cyclization cyclization selectively.