The application of aromaticity and antiaromaticity to reaction mechanisms
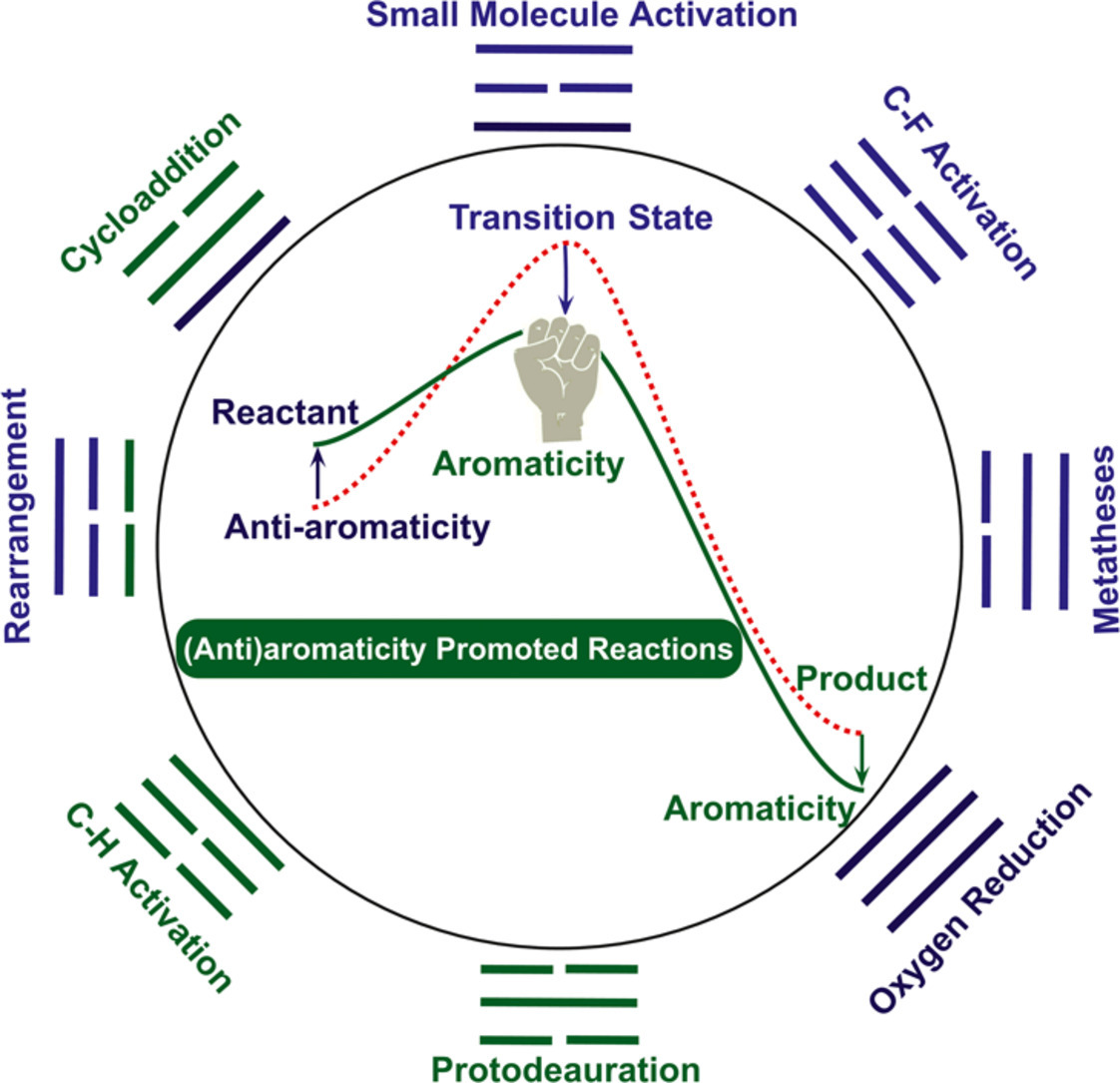
Aromaticity, in general, can promote a given reaction by stabilizing a transition state or a product via a mobility of π electrons in a cyclic structure. Similarly, such a promotion could be also achieved by destabilizing an antiaromatic reactant. However, both aromaticity and transition states cannot be directly measured in experiment. Thus, computational chemistry has been becoming a key tool to understand the aromaticity-driven reaction mechanisms. In this review, we will analyze the relationship between aromaticity and reaction mechanism to highlight an importance of density functional theory calculations and present it according to an approach via either aromatizing a transition state/product or destabilizing a reactant by antiaromaticity. Specifically, we will start with a particularly challenging example of dinitrogen activation followed by other small-molecule activation, C-F bond activation, rearrangement, as well as metathesis reactions. In addition, antiaromaticity-promoted dihydrogen activation, CO2 capture, and oxygen reduction reactions will be also briefly discussed. Finally, caution must be cast as the magnitude of the aromaticity in the transition states is not particularly high in most cases. Thus, a proof of an adequate electron delocalization rather than a complete ring current is recommended to support their relatively weak aromaticity in the transition states.
https://www.sciencedirect.com/science/article/pii/S2667325823001176