Theoretical studies on structures and spectroscopic properties of nitryl halogenides
Submitted by Jun Zhu on Thu, 10/31/2013 - 16:53
Authors:
Jun Zhu, Zexing Cao,* Qianer Zhang
Journal:
Acta Chim. Sin.
Year:
2002
Volume:
60
FirstPage-LastPage:
1040-1044
TOC:
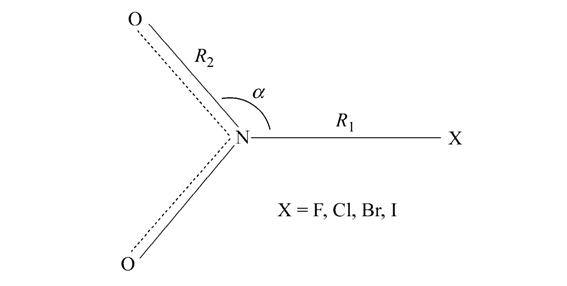
Abstract:
Density functional theory with the B3LYP functional is used to calculate the equilibrium geometries and harmonic vibrational frequencies of nitryl halogenides XNO2 and XONO (X = F, Cl, Br, I). Stabilities and isomerizations of these isomers are investigated. Dissociation energies of the X-N bond in XNO2 are predicted at the B3LYP/6-311G* and QCISD(T)/ce-pvTZ levels. The electronic transition energies of the most stable XNO2 species have been estimated by time-dependent B3LYP calculations. The electron promotion of a nonbonding electron of the halogen atom X in XNO2 into a pi* orbital on the NO2 moiety, i.e., the n-->sigma* electron excitation, is responsible for the photodissociation of the X-N bond.